ANN ARBOR, Mich. — Despite the jackhammer-like rhythm of a mechanical ventilator, Alicia Moreno had dozed off in a chair by her 1-year-old’s hospital bed, when a doctor woke her with some bad news: The common stool softener her son, Anderson, was given months earlier had been contaminated with the bacterium Burkholderia cepacia.
Suddenly, Anderson’s rocky course made medical sense. B. cepacia was the same unusual bacterium mysteriously found in the boy’s respiratory tract, temporarily taking him off the list for a heart transplant. The same bacterium resurfaced after his transplant and combined with a flu-like illness to infect his lungs. He’s been on a ventilator ever since.
The tainted over-the-counter medicine, docusate sodium, routinely prescribed to nearly every hospitalized patient to avert constipation, caused Anderson to suffer “serious and dangerous life-threatening injuries,” a lawsuit filed by his family alleges. The drug was eventually recalled, but only after a Texas hospital staff noticed an uptick in B. cepacia infections, prompting a six-month investigation that led back to the tainted drug and its Florida manufacturing plant.
“Something that was supposed to help him hurt him,” Alicia Moreno said.
Since the start of 2013, pharmaceutical companies based in the U.S. or abroad have recalled about 8,000 medicines, comprising billions of tablets, bottles and vials that have entered the U.S. drug supply and made their way to patients’ medicine cabinets, hospital supply closets and IV drips, a Kaiser Health News investigation shows. The recalls represent a fraction of the medicines shipped each year. But the flawed products contained everything from dangerous bacteria or tiny glass particles to mold — or too much or too little of the drug’s active ingredient.
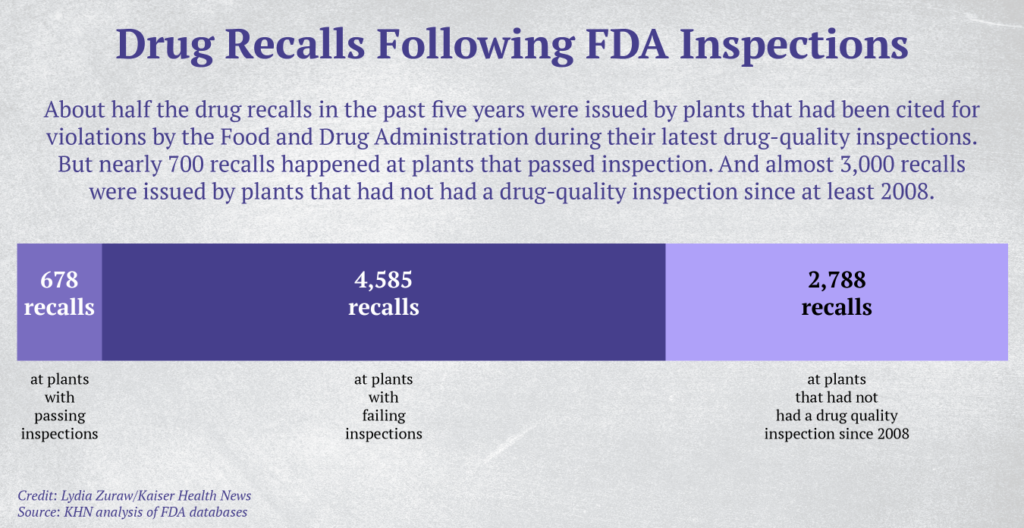
Over the same period, 65 drug-making facilities recalled nearly 300 products within 12 months of passing a Food and Drug Administration inspection — as was the case with the stool softener, according to a KHN analysis of recall notices and inspection records kept by the FDA.
Those recalls included more than 39,000 bottles of the HIV drug Atripla laced with “red silicone rubber particulates,” nearly 37,000 generic Abilify tablets that were “superpotent,” and nearly 12,000 boxes of generic Aleve (naproxen) that were actually ibuprofen, according to the recall data KHN examined.
The medicine alleged to have sickened Anderson Moreno seriously infected at least 63 other people in 12 states, according to reports by the FDA and Centers for Disease Control and Prevention. The drug was made at a PharmaTech plant in Broward County, Fla. That same plant passed an FDA inspection even while it was making bacteria-laced products, according to a KHN review of the inspection records.
PharmaTech did not respond to KHN’s requests for comment. A lawyer for the drugmaker filed a motion to dismiss the lawsuit in April, but it was not granted. In follow-up court records, PharmaTech has denied claims against it.
Like other FDA commissioners before him, Scott Gottlieb has called his agency’s drug oversight program the “gold standard” for safety and effectiveness.
But veteran industry consultant John Avellanet, who has trained FDA inspectors, questions how effective the FDA’s drug plant inspections actually are. “It’s so easy” for FDA inspectors to miss things because they’re working with confusing regulatory terms and standards that are often decades out of date, Avellanet said.
Just how often people are sickened or die from tainted drugs is next to impossible to determine. No government agency tracks cases unless they’re linked to a major outbreak among hospital patients. And sudden, seemingly random illnesses in disparate places are notoriously hard to link to a tainted drug. That’s in part because drugmakers don’t have to divulge which products are made in which manufacturing plants, since that is regarded as proprietary information.
The result: Even someone who buys drugs for a major hospital can’t track down where a potentially dangerous product came from, said Erin Fox, who purchases medicines for University of Utah Health hospitals.
“Patient safety should come first,” she said, adding that the KHN analysis indicates “our drug quality is probably not what we think it is,” and calling it a “scary” reality. “Something does need to change if this is happening this many times and we’re having patients receiving contaminated products.”
The FDA declined to be interviewed for this story, but responded to written questions.
“While the FDA would prefer that no drug be distributed that later is recalled, we do not think that a recall indicates a failure of FDA inspection and surveillance programs,” FDA spokesman Jeremy Kahn said in an email. He said inspectors “may not uncover all issues or practices that may eventually result in a problem leading to a recall” and that “not all recalls are the result of poor manufacturing practice.”
The PharmaTech Story
“Lucky fin, lucky fin, lucky fin,” Alicia Moreno, 30, cheered as she untangled her now 3-year-old son’s stroke-weakened arm from a sweater and his portable ventilator in the back seat of the car for yet another four-hour drive to see doctors in Ann Arbor, Mich. In the Disney movie “Finding Nemo,” Nemo’s father calls the young fry’s smaller fin his “lucky fin.”
While her husband drives, Alicia pulls out a clear plastic case of syringes and watches the clock on the dashboard. Anderson needs about two dozen different medicines every 24 hours, and Alicia administers them via a port in his belly at designated times.
Alicia spends the day with Anderson.
It wasn’t always like this. Anderson appeared healthy until his 6-month checkup in May 2016, his mother said. Partway through the exam, the Morenos rushed their baby to a nearby hospital and learned he was in heart failure and would need a transplant to survive. That’s where he received the tainted stool softener, his lawyers allege. The hospital where Anderson eventually received his transplant confirmed via email that Anderson tested positive for the same strain of B. cepacia involved in the outbreak traced back to PharmaTech’s contaminated drug.
In July, according to the family, Anderson started to have difficulty breathing and his temperature spiked to 106 degrees, which landed him in the ICU, where doctors and nurses packed him with ice and rushed to find the cause. Their tests turned up positive for B. cepacia, a bacterium found in untreated water that doesn’t typically make healthy people sick. Anderson’s status on the transplant list was put on hold, and his heart condition worsened. He was placed on a machine that transferred blood outside his body, oxygenated it and pumped it back in.
Anderson finally got a heart transplant in November 2016, but four days after doctors closed his chest, his fever was back and his lungs kept getting worse, requiring more complicated machinery. Tests came back positive for a flu-like virus and B. cepacia, according to the hospital.
“Where did he get it?” his parents pleaded. At the time, no one knew.
How Tainted Drugs Slip Through the Cracks

The FDA is supposed to inspect all factories, foreign and domestic, that produce drugs for the U.S. market. But a KHN review of thousands of FDA documents — inspection records, recalls, warning letters and lawsuits — offers insight into the ways poorly manufactured or contaminated drugs reach consumers: Inspectors miss serious hazards. Drugmakers fail to meet standards even after the FDA has taken enforcement action. Hundreds of plants haven’t been inspected for years, if ever.
Last July, for example, the FDA announced the first of many voluntary recalls of the blood pressure medicine valsartan because some tablets contain a cancer-causing impurity called N-nitrosodimethylamine (NDMA). They would later find a similar carcinogen, N-nitrosodiethylamine (NDEA), in valsartan pills. Over the prior two years, investigators had detected worrisome problems in two overseas factories involved in the manufacturing of the drug.
[protected-iframe id=”d01ad9e2d963ca21056cb18e097c1902-7618883-97277977″ info=”https://abcnews.go.com/video/embed?id=60156815″ width=”640″ height=”360″ style=”border: none;” scrolling=”no”]In 2017, FDA investigators found rust, chipping paint and deteriorating equipment at a plant run by Zhejiang Huahai Pharmaceutical Co. in Zhejiang, China. Plant staffers weren’t properly testing and investigating “anomalies” in their drugs, dismissing problematic test results, the FDA said at that time. Inspectors also found “black metallic particles” and other problems in some unidentified drugs.
The FDA inspected the plant in July 2018 after complaints about NDMA from a facility further down the drug supply chain. The FDA put the facility on import alert in late September and issued a warning letter in November detailing deficiencies, including “Failure of your quality unit to ensure that quality-related complaints are investigated and resolved.”
At a facility of Hetero Labs in India, in 2016, FDA inspectors found colored and white residue in components, some of the factory’s tablets were twice as thick as others, and employees were shredding documents in the middle of the night. The FDA issued a warning letter to the company as a result of the inspection.
Plants making drugs for U.S. consumers are supposed to be inspected every few years, according to a risk-based system. However, in the past decade more than 2,500 facilities, both foreign and domestic, have gone more than five years without an FDA drug-quality inspection, a KHN analysis found. The FDA has no drug-quality inspection records over the past decade for more than 1,200 domestic plants and nearly 400 foreign plants, excluding those that make animal drug products, according to the analysis. Gottlieb said in December that he hopes to clear the backlog of uninspected drug facilities by the end of September 2019.
At best, the inspections are a snapshot in time, and involve looking at processes rather than evaluating the drugs themselves, said drug-quality specialist Dinesh Thakur, who has worked for drugmakers. The inspections might take place while the facility is making only one of a dozen or so drugs that it usually manufactures.
“The implicit assumption … is that if the [manufacturing] processes are sound, the product will be of good quality,” said Thakur, who raised the alarm about quality-control problems at generics drugmaker Ranbaxy, resulting in a 2013 guilty plea and a $500 million settlement. “Your data tells us this is not true.”
Many inspections, he said, are “stage-managed,” so that factories pass on the appointed day, but “once the inspectors leave, it’s a completely different story.”
David Gortler, a former FDA medical officer, said most drug plant inspections involve looking over paper records and trusting that they’re real, instead of randomly testing medicines.
“Anybody can write down anything on a piece of paper,” said Gortler, who is now a consultant at FormerFDA.com. He added that FDA inspectors aren’t reprimanded — or even told — if they’ve passed a plant that issued a recall shortly thereafter.
A sign on the Morenos’ front door asks visitors not to enter if they have recently been sick. [khn_two_photos photo-first=”897303″ photo-second=”897304″]
A Lucky Break Solves A Mystery
The contaminated stool softener alleged to have sickened Anderson Moreno was one of many drugs recalled by plants shortly after they passed an FDA inspection. The bacteria was detected only after an outbreak of disease — and after a good deal of medical sleuthing.
More than 1,000 miles away from Anderson’s ICU bed in Michigan, staff at Texas Children’s Hospital’s pediatric ICU in Houston had diagnosed three cases of B. cepacia in one week in February 2016, according to a 2017 medical journal article published in Infection Control and Hospital Epidemiology. It was odd because there had been no cases the previous year.
Hospital staff members embarked on a months-long investigation and by July had identified 24 victims, whose median age was under 2 years old. Patients had the same strain of the bacteria in their blood, their respiratory tracts, their urine or their stool, according to the article.
Samples matched the bacteria found in liquid docusate, the stool softener, the researchers wrote.
The hospital alerted the CDC and other public health officials of its findings. The CDC would eventually identify 63 confirmed and 45 suspected serious B. cepacia infections in 12 states tied to the contaminated drug.
A 36-day FDA inspection of PharmaTech in Davie, Fla., that ended Aug. 9, 2016, revealed that the bacteria was in water used to clean equipment and make liquid products. FDA inspectors concluded that the bacterium made it into the facility’s drugs starting in 2015 and was still present in the water.
Anderson was treated with the stool softener in May 2016. His parents filed suit in September 2017 in PharmaTech’s home of Broward Country, Fla., against the drugmaker and others in the drug supply chain, alleging the drug was contaminated and caused him grievous damage. PharmaTech, which did not return KHN’s requests for comment, unsuccessfully filed a motion to dismiss and has denied all charges in a subsequent filing.
A 9-month-old girl in Pittsburgh who had received the stool softener died on May 4, 2016, according to a lawsuit her family filed in July 2017 in the U.S. District Court for the Western District of Pennsylvania. Her mother learned about the drug recall by chance and asked the hospital whether her deceased daughter received the tainted drug, her lawyer told KHN. The family filed charges against PharmaTech and others in the drug supply chain in a wrongful death lawsuit. The court rejected PharmaTech’s motions to dismiss and strike, and the drugmaker denied liability in a subsequent filing. In November 2017, a lawyer representing PharmaTech in that wrongful death case told the Orlando (Fla.) Sun Sentinel that it will defend itself against the allegations and couldn’t comment further “because of the ongoing nature of litigation.”
According to federal records, FDA inspectors had a chance to catch the contamination during their March 2016 inspection, but the PharmaTech plant passed with no citations. PharmaTech CEO Ray Figueroa saluted the inspection results in a press release, calling it “a testimony to PharmaTech’s commitment to world-class quality.”
How Things Can Go Wrong
The FDA has issued thousands of enforcement actions against drug plants over the years, citing safety violations, issuing warning letters and blocking imports from certain foreign plants. In rare cases, the FDA can also seize drug products and has done so 23 times in the past decade. The last drug seizure was more than two years ago, according to FDA records.
In an emailed statement, FDA chief Gottlieb said the FDA is “taking new steps” to identify problems before they occur and it is “not shy” to use its powers to mitigate risks.
But the system can be stymied or gamed and the FDA’s enforcement abilities are limited. For instance, it doesn’t have the power to issue a mandatory recall, and manufacturing citations don’t come with fines.
Many cases come to light only when a whistleblower sounds an alarm.
Thakur, the Ranbaxy whistleblower, said officials in other countries sometimes tip off plants about “surprise” FDA inspections. And FDA inspectors often have to rely on translators hired by the drug companies, said Avellanet, who has been a drug facility inspection consultant for more than 20 years.
At Nippon Fine Chemical in Japan, employees stood “shoulder-to-shoulder” to keep an FDA official out in December 2015, according to an enforcement letter sent to the plant and published online.
Less than a year later, Vikshara Trading & Investments Ltd. in India allegedly faked a worker strike to block the entrance to the plant, according to an FDA enforcement document that described the manufacturer’s “false statements.” When inspectors were eventually allowed in, the lights were kept off.
“Our investigator had to perform parts of the walkthrough in the dark, using a flashlight,” the FDA warning letter reads, adding that an unidentified powder was “scattered” and “caked on the floor” in production areas and detected on finished drug products.
Two former employees have filed a whistleblower suit against Gilead Sciences, alleging it lied to the FDA about using a drug-manufacturing facility in South Korea, when it was actually using an unregistered facility in China. According to the civil complaint filed in September 2014 in U.S. District Court for the Northern District of California, the ingredient produced at Synthetics China and used in HIV drugs Truvada and Atripla contained “glass-like shards,” “black rubber-like particles,” “plastic-like particles,” “small stone or pebble-like particles” and “metal shards.”
The whistleblowers alleged Gilead’s Alberta, Canada, plant was tasked with sieving contaminants and helping to conceal where the ingredient was made.
They said one batch of the ingredient was contaminated with arsenic, chromium and nickel. Another had a dangerous bacterium called Bacillus cereus, according to the whistleblowers’ suit. Still, Gilead released the product and didn’t initiate a recall, the whistleblowers alleged.
Years after the whistleblowers stopped working for Gilead, the drugmaker issued two voluntary recalls of HIV drugs in 2014, about seven months apart. Both recalls cited contamination with red silicone rubber particulates.
Gilead declined to comment. Gilead has fought the lawsuit, alleging that since the government knew of the allegations and did not penalize it by denying drug approvals or payments, the suit could not move forward. In 2015, a federal judge dismissed the case, but a panel from the 9th District Court of Appeals reversed that decision in 2017. Now the Supreme Court may hear it; in April 2018, it invited the solicitor general to file a brief, “expressing the views of the United States.” The Justice Department filed a brief in November, saying pursuing the lawsuit is “not in the public interest.”
Since the FDA has little power to force a drugmaker to fix problems or issue recalls, FDA inspectors often flag the same violations again and again. A KHN analysis found that over the past decade 70 drug plants — most of them domestic — were penalized for the same violation at least four times. And more than a third of those plants has issued a recall at some point.
Altaire Pharmaceuticals in New York was cited five times by FDA inspectors for inadequate “procedures for sterile drug products.” In 2013, it recalled 363,746 bottles of generic eye drops sold at CVS, Target and Walmart over sterility concerns — namely mold — because the preservative in the product “may not be effective” through the expiration date. Overall, Altaire was told to correct 15 violations at least twice.
KHN attempted to contact Altaire Pharmaceuticals, but the company did not reply.
Kept In The Dark
About a year after the initial PharmaTech recall in 2016, the FDA announced another recall for the same drugs and the same bacterium: B. cepacia. When Erin Fox saw the second recall, she thought it was a mistake. The alert said to avoid drugs made by PharmaTech under several labels “and possibly [products from] other companies.” What other companies? Fox wondered. How could they not know which ones?
Doctors at the hospital asked Fox to remove all PharmaTech-made products from the shelves, but because of lax labeling laws, she said, she couldn’t be sure which those were. Drug labels need to include only the manufacturer, the packer or the distributor — not all three — so the doctors suggested she call PharmaTech and ask what else it manufactures and for whom.
“Of course,” Fox said, “they wouldn’t tell us.”
Methodology
To analyze the inspections and recalls of plants that manufacture drugs, KHN started with two Food and Drug Administration databases of drug recalls: one at OpenFDA, and one on the FDA’s recall data dashboard. The first provided details about drugs, dates and quantities recalled, and the second provided a recalling plant ID, called an FEI. We used them both to create a more complete recall database.
The FEIs served as a bridge between the recalls data and two inspection data tables. Both tables contained inspection dates and purposes, but one listed inspection grades and the other contained a list of citations. Combining inspection and recall databases allowed us to find the most recent inspection at each plant that preceded a recall, and to determine its grade. It also allowed us to count repeat citations and determine whether plants that received them ever initiated a recall.
To determine whether plants had not been inspected in the past decade, we compared our inspections data to the Drug Establishments Current Registration Site database, which contains all registered operating plants. We excluded plants that made products for animals and those that didn’t explicitly “manufacture” drugs, according to the database. The FDA has said there may be a delay in adding inspections to its database after they are completed.
Our data is current as of early October 2018. We included only inspections categorized as “drug quality assurance” inspections throughout the analysis.
KHN’s coverage of prescription drug development, costs and pricing is supported in part by the Laura and John Arnold Foundation.


